Sickle Cell Anemia
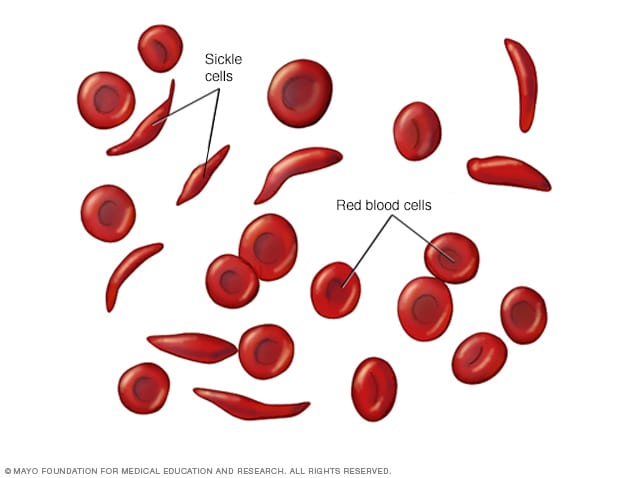
Sickle cell anemia is a genetic disease prevalent in human populations in Africa and other warmer regions of the world. A mutated allele of a gene coding for β - globin subunit of hemoglobin, the oxygen carrying pigment, causes an abnormality of the hemoglobin molecules. The presence of this abnormal hemoglobin in red blood cells changes the shape of the RBCs to curve as a sickle from its disk-shape. Individuals with this disorder have fewer numbers of RBCs, and hence develop anemia. This is because the sickled RBCs breakdown prematurely. The mutation substitutes glutamic acid at a particular place in the primary structure of the β-globin with valine which results in abnormal folding of hemoglobin. The mutated allele is codominant, which means both normal β globin and mutated β globin are produced in individuals who are the heterozygous for this locus. Therefore, they have both good and bad hemoglobin and hence both normal and sickled RBCs are present. They are normally healthy and are the carriers of the mutant allele. Since the mutated alleles cause severely detrimental effects in homozygous individuals, normally they should have been eliminated from the human population by natural selection. However, there is prevalence of malaria in warmer countries like in Africa; the heterozygous individuals will survive malaria attacks better than individuals with homozygous wild type alleles. This is because, the malaria parasite cannot survive in sickled RBCs. Therefore, at heterozygous individuals, parasite density remains at a lower level.
Comments
Post a Comment